

Abstract
Bosutinib is a second-generation Abl tyrosine kinase inhibitor (TKI) that was developed for the treatment of patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML). Mainly metabolized by cytochrome P450 (CYP) 3A4, this TKI is a substrate for ATP-binding cassette (ABC) transporters, such as P-glycoprotein, encoded byABCB1, and breast cancer resistance protein (BCRP), encoded by ABCG2. Therefore, polymorphisms in CYP3A4and ABCB1or ABCG2may affect the pharmacokinetics of bosutinib. When patients with CML are administered TKI therapy, therapeutic drug monitoring (TDM) may represent a new strategy for optimizing the dosage to achieve faster or deeper responses or reduce the toxicity of the TKI therapy, if the target plasma concentration of bosutinib is identified.
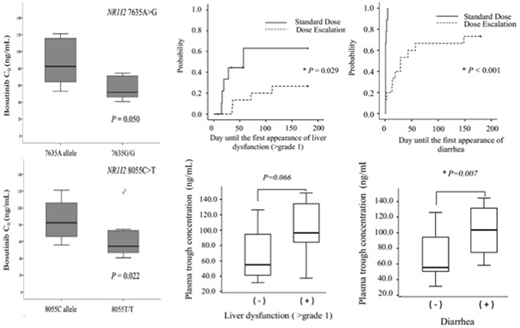
We first investigated the effects of polymorphisms in CYP3A4(20230G>A), ABCB1(1199G>A, 1236C>T, 2677G>T/A, 3435C>T), ABCG2(421C>A), and NR1I2(7635A>G, 8055C>T) on the mean plasma trough concentrations (C0) of bosutinib at steady-state in 17 Japanese patients with CML who were receiving 300 mg/day bosutinib. Bosutinib C0values were monitored using high-performance liquid chromatography (HPLC-UV). No significant differences in bosutinib C0among patients with genotypes for CYP3A4,ABCB1, andABCG2polymorphisms were found. However, bosutinib C0in patients with the NR1I27635G/G or 8055T/T genotype was significantly lower than that in patients with the 7635A or 8055C allele, respectively (P=0.050,P=0.022). Patients with the NR1I27635G/G or 8055T/T genotype may have more active ABC efflux drug transporters and may consequently have a lower bosutinib C0. Therefore, information on the NR1I2 genotype may be useful to achieve the optimal systemic exposure of bosutinib.
Next, we conducted a prospective clinical study to investigate the exposure-toxicity relationship of bosutinib and to identify the target trough plasma concentration (C0). The toxicity and C0of bosutinib in patients with CML were monitored every 2 weeks for the first 3 months of treatment, and subsequently once per month during the 6 months after they started receiving 500 mg/day of the standard dose (the SD group, n=10) or started at 100 mg/day with increases of 100 mg every 2 weeks for dose escalation (the DE group, n=15). Nine out of 10 patients in the SD group were unable to continue bosutinib therapy without interruption owing to adverse events, compared with 2 out of 15 patients in the DE group. The median time until liver dysfunction was 28 days in the SD group, and 53.5 days in the DE group. Meanwhile, the median time until diarrhea was 1 day in the SD group, and 19 days in the DE group. The cumulative dose of bosutinib was comparable between the SD and DE groups (51,700 mg vs. 53,550 mg), and the median C0was 63.7 ng/mL and 63.0 ng/mL, respectively. Liver dysfunction and diarrhea were prevalent in quartile 4 of C0(>91.0 ng/mL), as calculated by the total C0distribution.
In conclusion, information on the NR1I2genotype may be useful to achieve the optimal systemic exposure to bosutinib. In addition, the DE regimen more effectively avoided treatment interruption. The daily dose of bosutinib should be adjusted based on target C0 (63-91 ng/mL) to increase efficacy and avoid adverse events by TDM in general practice.
Takahashi:Novartis: Research Funding, Speakers Bureau; Otsuka: Research Funding, Speakers Bureau; Pfizer: Research Funding, Speakers Bureau; Bristol-Myers Squibb: Research Funding, Speakers Bureau.

